Sodium-ion batteries (SIBs) are attractive for large-scale energy storage due to the abundance and low cost of sodium resources. However, cathode materials with single transition metal redox have limited capacity, hindering their further application. Recently, layered transition metal oxides have shown high specific capacity owning to the redox of transition metals and oxygen and have emerged as a new path to optimize the electrochemical performance of cathodes. Therefore, it is important to investigate the special structure and evolution mechanism in anionic redox reaction. In this study, the formation mechanism and structural regulation in anionic redox reaction are presented. This review is expected to offer a reference for designing high-performance cathode materials in SIBs.
ZHENG Wei. Modulating anionic redox reaction in layered transition metal oxides for sodium-ion batteries. Energy Storage Science and Technology[J], 2020, 9(5): 1416-1427 doi:10.19799/j.cnki.2095-4239.2020.0169
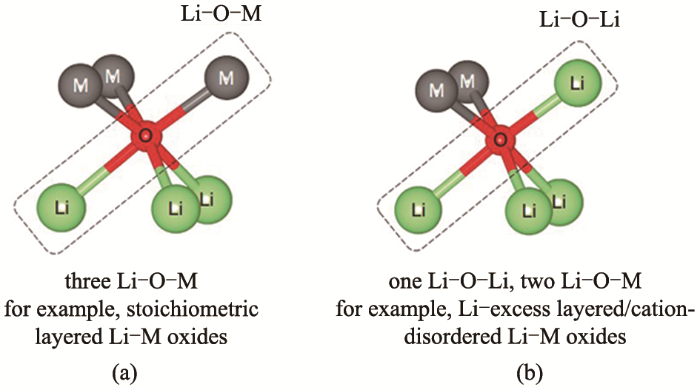
Fig.1
The main structure of layered transition metal oxides for sodium-ion batteries[7]
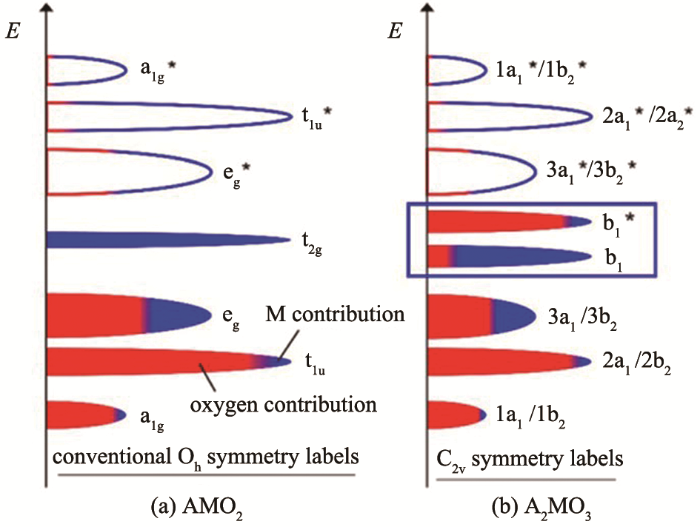
2 阴离子氧化还原反应的发展历程和机理
早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量。1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22]。在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27]。2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量。同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]。
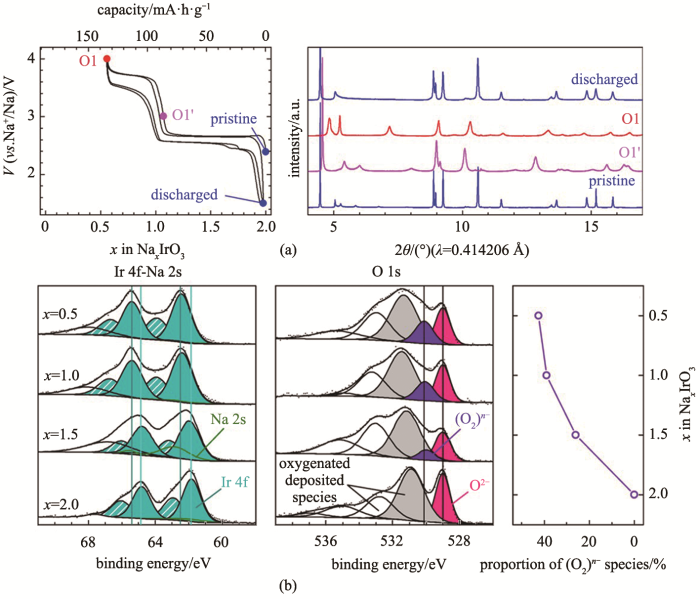
Fig.4
(a) charge and discharge profile for Na2IrO3 and corresponding XRD at different voltage; (b) XPS for Ir-4f and O-1s at different voltage states and the corresponding ratio of (O2)n-[35]
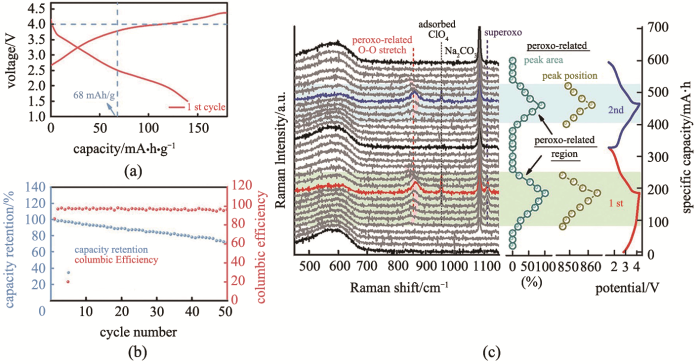
Fig.5
(a) charge and discharge in the first cycle; (b) the cycle performance and coulombic efficiency; (c) the charges of Raman spectra and the ratio of peroxides for Na1.2Mn0.4Ir0.4O2[36]
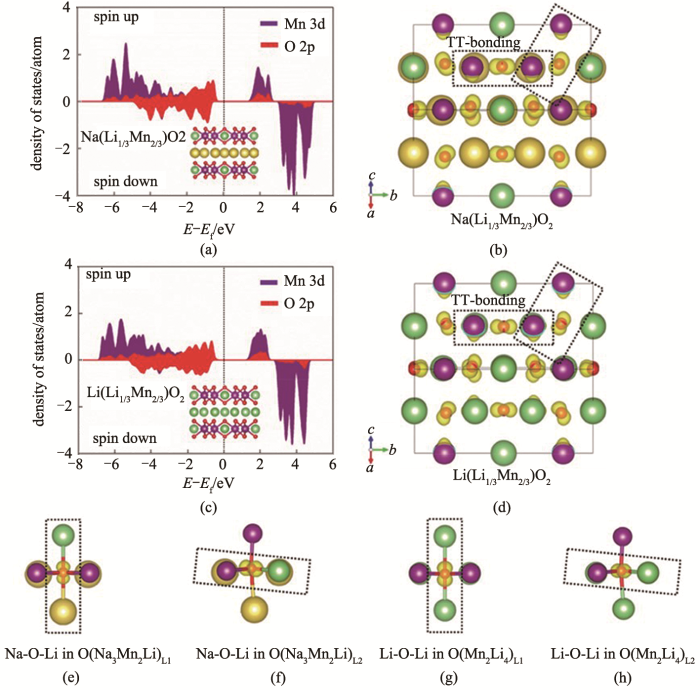
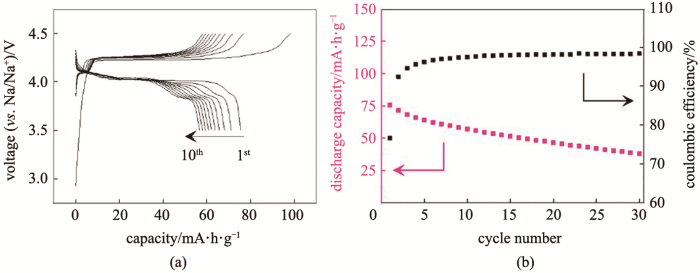
当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型。Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6)。和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用。Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层。如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%。通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量。作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散。除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39]。最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关。如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞。与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小。通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位。在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件。在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞。
Fig.8
First-cycle voltage curves for (a) Na0.75[Li0.25Mn0.75]O2 and (c) Na0.6[Li0.2Mn0.8]O2; (b) structural model of P2-type Na[TM]O2 with no in-plane ordering; (e) structural model of ,corresponding XRD and ADF-STEM profile with [010] direction in Na0.75[Li0.25Mn0.75]O2; (d) structural model of, corresponding XRD and ADF-STEM profile with [010] direction in Na0.6[Li0.2Mn0.8]O2
Fig.10
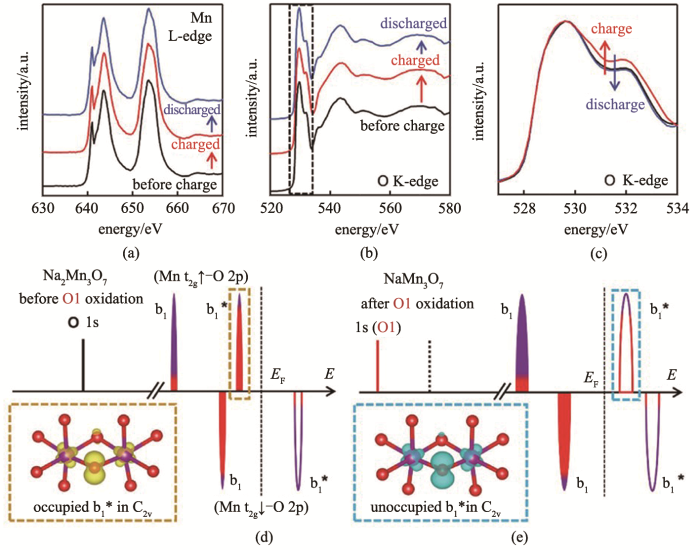
Ex situ X-ray absorption spectra for (a) Mn L3-edge; (b) O K-edge; (c) enlarged part of X-ray absorption spectra for O K-edge; (d) Schematic illustration of the electronic structures of Na2Mn3O7 and (e) NaMn3O7[44]
Fig.11
P2-Na2/3Mn7/9Zn2/9O2 (a) charge-discharge cycles with the derivative plot as inset and capacity retention plot and rate capability signature as inset; (b) PDOS profile and the inset show the PDOS of Na2/3Mg1/3Mn2/3O2 for comparison;
Fig.12
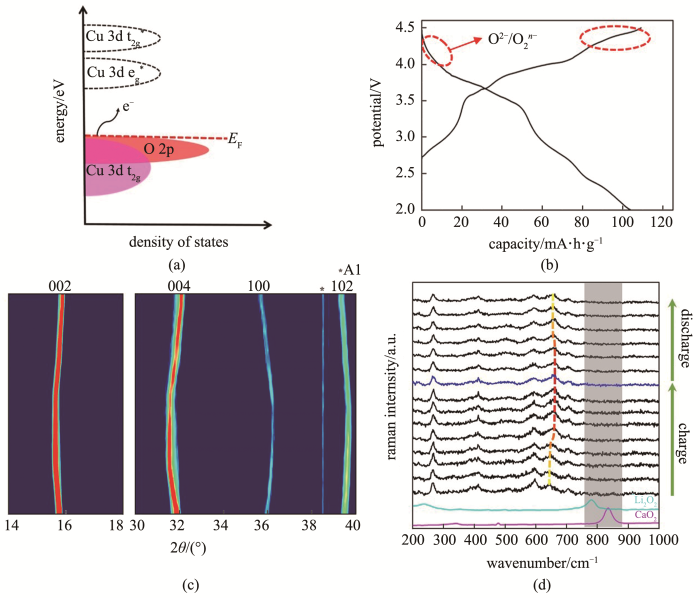
(a) schematic molecular orbital and electronic states; (b) charge and discharge curves; (c) in-situ XRD; (d) in-situ Raman during the first cycle for Na2/3Mn0.72Cu0.28O2
The main structure of layered transition metal oxides for sodium-ion batteries<sup>[<xref ref-type="bibr" rid="R7">7</xref>]</sup>Fig.12 阴离子氧化还原反应的发展历程和机理
早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
... [7]Fig.12 阴离子氧化还原反应的发展历程和机理
早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Electrode materials for rechargeable sodium-ion batteries: Potential alternatives to current lithium-ion batteries
0
2012
Room-temperature stationary sodium-ion batteries for large-scale electric energy storage
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries
In situ structural and electrochemical study of Ni1-xCoxO2 metastable oxides prepared by soft chemistry
1
1999
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides
0
1997
Identification of cathode materials for lithium batteries guided by first-principles calculations
1
1998
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Oxygen contribution on Li-ion intercalation-deintercalation in LiCoO2 investigated by O K-edge and Co L-edge X-ray absorption spectroscopy
1
2002
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Direct In situ observation of Li2O evolution on Li-rich high-capacity cathode material, Li[NixLi(1-2x)/3Mn(2-x)/3]O2(0<x<0.5)
0
2014
Fundamental interplay between anionic/cationic redox governing the kinetics and thermodynamics of lithium-rich cathodes
0
2017
Soft X-ray absorption spectroscopic and Raman studies on Li1.2Ni0.2Mn0.6O2 for lithium-ion batteries
0
2012
Direct visualization of the reversible O2-/O- redox process in Li-rich cathode materials
1
2018
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Visualization of O-O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries
1
2015
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Performance and design considerations for lithium excess layered oxide positive electrode materials for lithium ion batteries
1
2016
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
High performance Li2Ru1–yMnyO3 (0.2≤y≤0.8) cathode materials for rechargeable lithium-ion batteries: Their understanding
1
2013
... 早在1990年,研究者在锂离子电池硫化物正极材料(如TiS3、FeS2)中发现,在电化学反应过程中S存在S2-/(S2)2-的可逆转变,可为正极材料提供容量.1999年,Tarascon等通过同步辐射以及一些磁性测试发现,高充电电压下的LixCoO2中具有氧的价态变化[20-22].在此之后,各种先进的测试手段如X射线光电子能谱分析(X-ray photoelectron spectroscopy,XPS)、X射线吸收近边结构分析(X-ray absorption near edge structure,XANES)、共振非弹性X射线散射 (resonant inelastic X-ray scattering,RIXS)、电子能量损失谱(electron energy loss spectroscopy,EELS)、拉曼等被用来研究氧在电化学反应过程中的氧化还原行为及转化机制[12, 23-27].2015年,Tarascon等[28]通过扫描透射电镜(scanning transmission electron microscope,STEM)实际观测到了充电过程中O—O键的缩短,证明了氧的价态变化可为材料贡献比容量.同时,研究人员通过各种实验和计算手段分析氧的反应和转化机制,并通过对结构的调控设计更多具有阴离子氧化还原反应的新材料[29-30]. ...
Charge compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen
... [35]<strong>(a) charge and discharge profile for Na<sub>2</sub>IrO<sub>3</sub> and corresponding XRD at different voltage; (b) XPS for Ir-4f and O-1s at different voltage states and the corresponding ratio of (O<sub>2</sub>)<i><sup>n</sup></i></strong><sup>-</sup><sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup>Fig.4
(a) charge and discharge in the first cycle; (b) the cycle performance and coulombic efficiency; (c) the charges of Raman spectra and the ratio of peroxides for Na<sub>1.2</sub>Mn<sub>0.4</sub>Ir<sub>0.4</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R36">36</xref>]</sup>Fig.5
当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型.Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6).和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用.Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层.如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%.通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量.作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散.除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39].最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关.如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞.与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小.通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位.在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件.在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞. ...
(a) charge and discharge in the first cycle; (b) the cycle performance and coulombic efficiency; (c) the charges of Raman spectra and the ratio of peroxides for Na<sub>1.2</sub>Mn<sub>0.4</sub>Ir<sub>0.4</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R36">36</xref>]</sup>Fig.5
当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型.Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6).和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用.Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层.如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%.通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量.作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散.除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39].最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关.如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞.与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小.通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位.在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件.在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞. ...
Manganese-based Na-rich materials boost anionic redox in high-performance layered cathodes for sodium-ion batteries
... [36](a) charge and discharge in the first cycle; (b) the cycle performance and coulombic efficiency; (c) the charges of Raman spectra and the ratio of peroxides for Na<sub>1.2</sub>Mn<sub>0.4</sub>Ir<sub>0.4</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R36">36</xref>]</sup>Fig.5
当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型.Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6).和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用.Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层.如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%.通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量.作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散.除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39].最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关.如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞.与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小.通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位.在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件.在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞. ...
... [36]Fig.5
当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型.Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6).和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用.Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层.如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%.通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量.作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散.除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39].最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关.如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞.与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小.通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位.在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件.在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞. ...
Rational design of Na(Li1/3Mn2/3)O2 operated by anionic redox reactions for advanced sodium-ion batteries
3
2017
... 当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型.Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6).和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用.Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层.如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%.通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量.作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散.除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39].最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关.如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞.与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小.通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位.在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件.在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞. ...
... [37](a) PODS; (b)ELF; (e)~(f) Na-O-Li configuration for Na(Li<sub>1/3</sub>Mn<sub>2/3</sub>)O<sub>2</sub>; (c)PODS; (d)ELF; (g)~(h) Li-O-Li configuration for Li(Li<sub>1/3</sub>Mn<sub>2/3</sub>)O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig.6
(a) charge and discharge profile for Na<sub>0.6</sub>Li<sub>0.2</sub>Mn<sub>0.8</sub>O<sub>2</sub>; (b) cycle performance for Na<sub>0.6</sub>Li<sub>0.2</sub>Mn<sub>0.8</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup>Fig.7
(a) charge and discharge profile for Na<sub>0.6</sub>Li<sub>0.2</sub>Mn<sub>0.8</sub>O<sub>2</sub>; (b) cycle performance for Na<sub>0.6</sub>Li<sub>0.2</sub>Mn<sub>0.8</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup>Fig.7
Structure-induced reversible anionic redox activity in Na layered oxide cathode
3
2018
... 当把过渡金属层中的过渡金属换成碱金属Li时,材料中便产生了Na-O-Li构型.Kim等[37]通过密度泛函理论(density functional theory,DFT)计算了Na(Li1/3Mn2/3)O2的偏分态密度图(partial density of states,PDOS)和局域密度函数图(electron localization function,ELF),并同Li(Li1/3Mn2/3)O2进行对比(图6).和Li(Li1/3Mn2/3)O2中氧的行为相似,PDOS图显示O-2p的电子态占据了更高的能级,比Mn-3d电子态更加靠近费米能级,电子可以从O中脱出,从而在钠离子脱嵌过程中O2-/O-的氧化还原可以贡献容量,而来源于Na-O-Li构型的O-2p轨道的非键态在氧的氧化还原反应中有关键作用.Rong等[38]报道了在P3-Na0.6Li0.2Mn0.8O2中的阴离子氧化还原反应,在该化合物中,Li取代部分的Mn的位置占据过渡金属层.如图7所示,材料在3.5~4.5 V的高电压区间,30 mA/g的电流密度下具有约100 mA·h/g的充电比容量,放电比容量为75 mA·h/g,30圈的循环后容量保持率为49.3%.通过XPS、XANES等测试表征,证明了在充电过程中氧直接参与到电荷补偿中,O2-/(O2)n-的氧化还原反应为材料提供容量.作者进一步对材料的容量衰减机理进行研究,发现材料容量衰减的原因有两个:第一,充电时材料发生堆叠层错,后无法复原,使得Na+无法重新嵌回到材料中;第二,高活性的O官能团和电解液发生反应,使得固体电解质界面层越来越厚,影响钠离子的扩散.除此以外,他们还报道了同类型材料Na0.72Li0.24Mn0.76O2中的阴离子氧化还原效应[39].最近,Bruce等[40]对上述两种化合物Na0.75Li0.25Mn0.75O2和Na0.6Li0.2Mn0.8O2进行更深入的研究,他们关注到材料电压迟滞和过渡金属层中Li/Mn的有序性相关.如图8所示,蜂窝型结构的P2-Na0.75Li0.25Mn0.75O2属于P63/mmc空间群,在充放电过程中具有严重的电压迟滞.与之不同,属于P21/c空间群、带状型结构的P2-Na0.6Li0.2Mn0.8O2在充放电过程中电压迟滞却很小.通过实验手段和计算手段的深入分析,作者指出碱金属从过渡金属层和碱金属层脱出后,过渡金属层含有一定量的空位.在蜂窝型结构的化合物中,只要两个Mn同时迁移到近邻的空位就能促进On-变成O2,从而使放电电压降低造成严重的电压迟滞;而在带状结构的化合物中,空位是分散的,Mn需要经过多次的迁移或者跳跃才能形成过渡金属空位的团簇,进而为氧气生成创造条件.在这种情况下,O2生成的条件更苛刻,从而促进了氧的稳定、抑制了电压迟滞. ...
... [38](a) charge and discharge profile for Na<sub>0.6</sub>Li<sub>0.2</sub>Mn<sub>0.8</sub>O<sub>2</sub>; (b) cycle performance for Na<sub>0.6</sub>Li<sub>0.2</sub>Mn<sub>0.8</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup>Fig.7
... [41](a) charge and discharge profile for Na<sub>2/3</sub>Mg<sub>0.28</sub>Mn<sub>0.72</sub>O<sub>2</sub>; (b) gas loss of Na<sub>2/3</sub>Mg<sub>0.28</sub>Mn<sub>0.72</sub>O<sub>2</sub> during electrochemical (de)sodiation<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig.9
... [44]<i>Ex situ</i> X-ray absorption spectra for (a) Mn L3-edge; (b) O K-edge; (c) enlarged part of X-ray absorption spectra for O K-edge; (d) Schematic illustration of the electronic structures of Na<sub>2</sub>Mn<sub>3</sub>O<sub>7</sub> and (e) NaMn<sub>3</sub>O<sub>7</sub><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup>Fig.10








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}